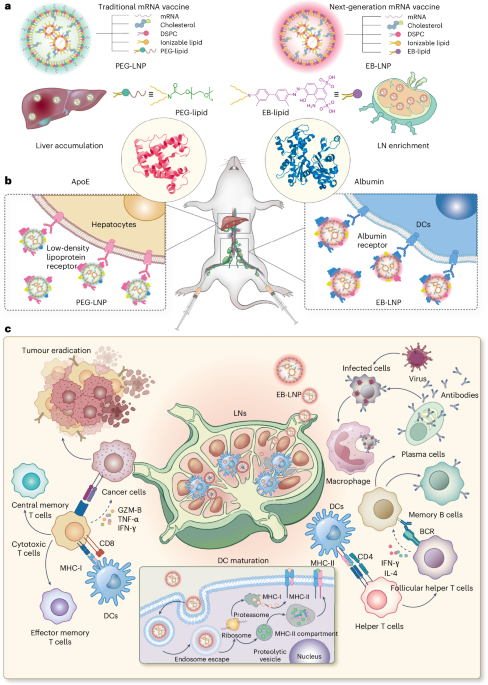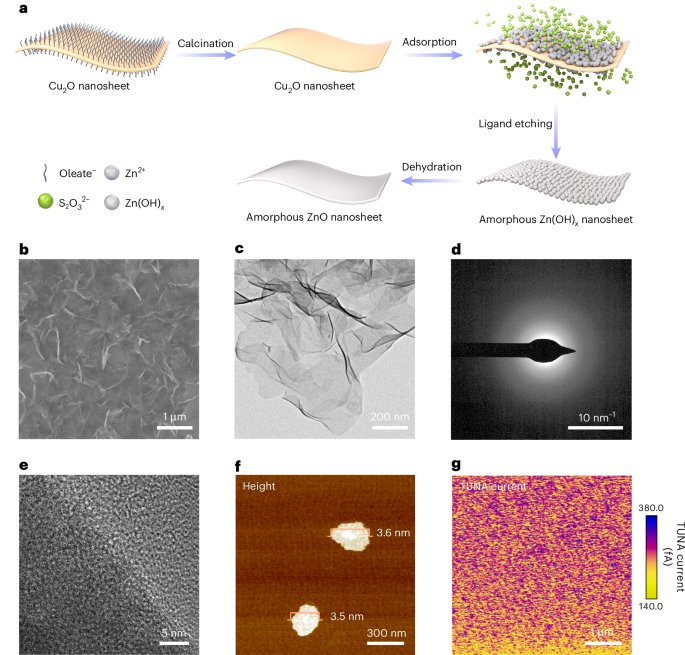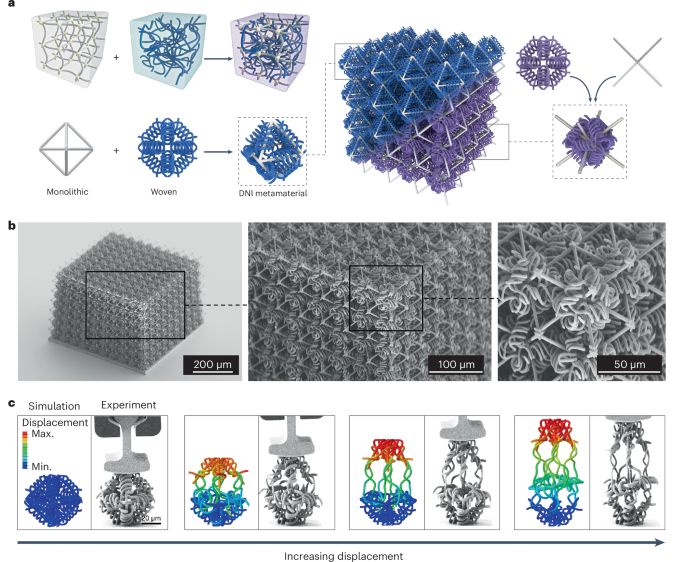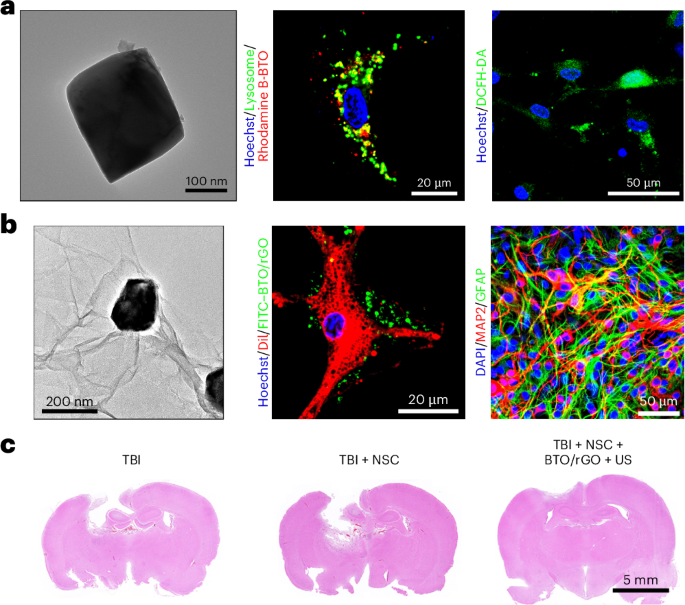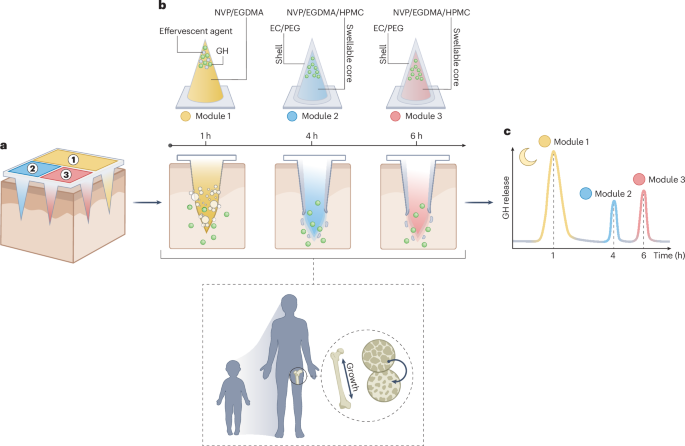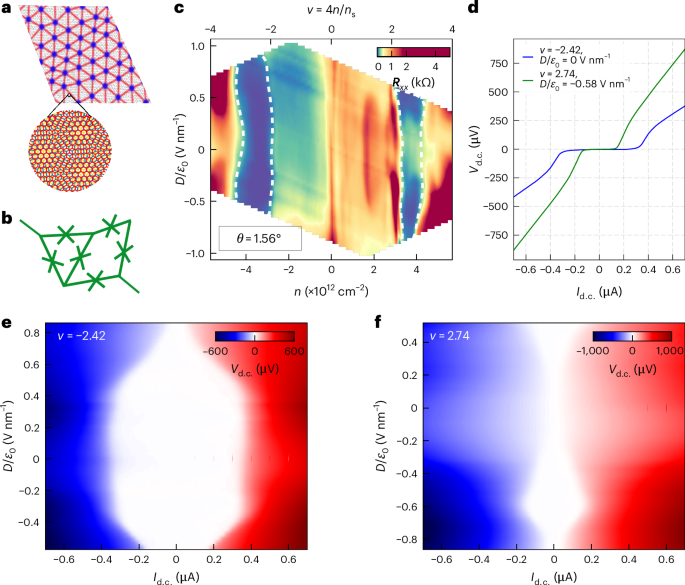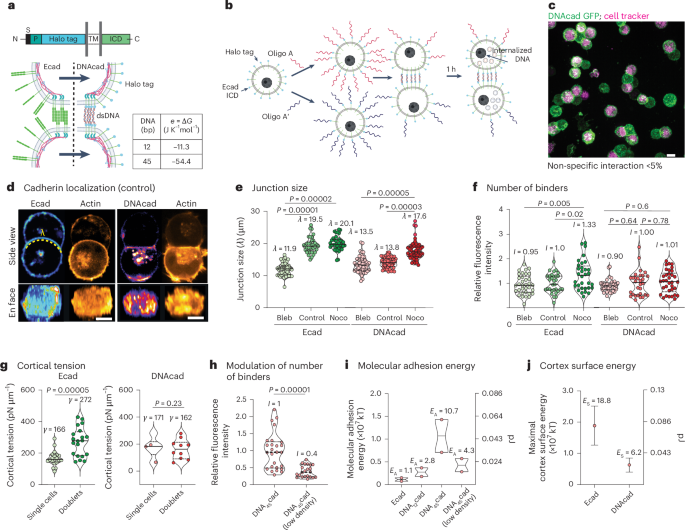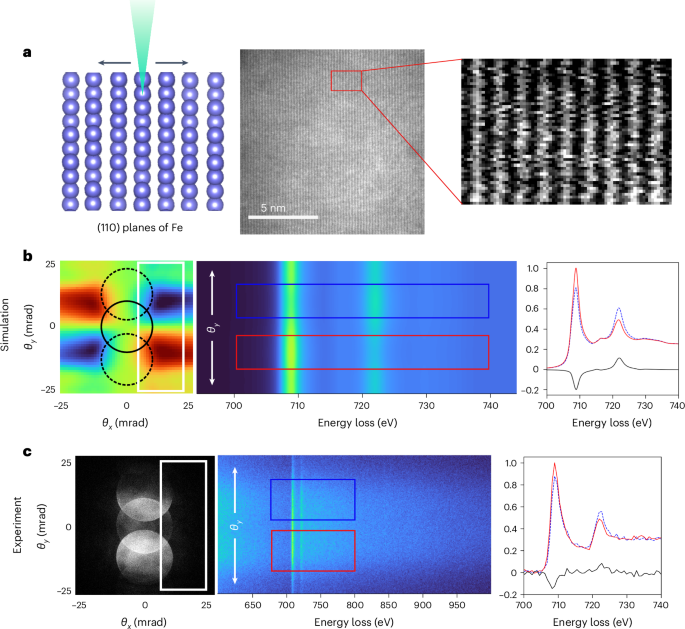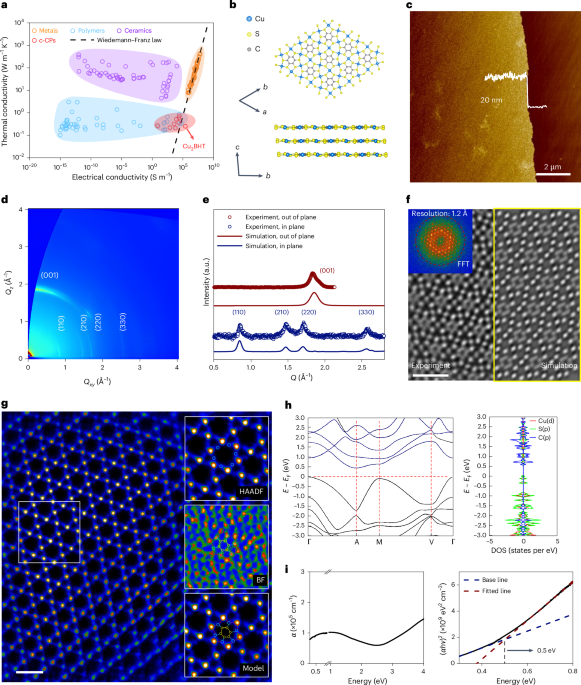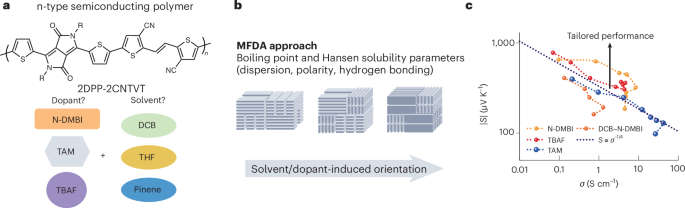
Mouse studies were approved by the Institutional Animal Care and Use Committee of The University of Texas, MD Anderson Cancer Center, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (IACUC protocol 00001002). All procedures involving the chick embryo model were approved by the University of Alberta Institutional Animal Care and Use Committee.
Cell culture and pharmacological inhibitors
Human HT-1080 fibrosarcoma cells, MDA-MB-231 breast cancer cells, A431 epidermoid carcinoma and human foreskin fibroblasts (HFF-1) cells were acquired from American Type Culture Collection. HOS cells were obtained from the NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH). HT-1080, MDA-MB-231, HOS, A431 and HFF-1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g l–1 glucose, l-glutamine and sodium pyruvate (Gibco) and supplemented with 10% heat-inactivated foetal bovine serum (Gibco, 16140071) and 1% penicillin/streptomycin (10,000 U ml–1; Gibco, 15140122).
BRC-196 breast cancer cells (kindly provided by Dr. Seagel at McGill University) were cultured in DMEM/F-12 containing 3.151 g l–1 of glucose, 15 mM of HEPES, l-glutamine and sodium pyruvate supplemented with 10% heat-inactivated foetal bovine serum (Gibco, 16140071), 10 nM of β-estradiol (Sigma, E8875), 0.4 µg ml–1 of hydrocortisone (MilliporeSigma, H0135), 5 ng ml–1 of heregulin-β1 (STEMCELL Technologies, 78071) and 4 µg ml–1 of insulin (Sigma, I9278). Cells were grown in an incubator maintained at 37 °C and 5% CO2, and subcultured every 2–4 days.
In select experiments, cells were treated with the following pharmacological agents and the corresponding vehicle controls. Reagents were obtained from Sigma-Aldrich unless otherwise noted: Y27632 (Y0503, 10 µM), hydroxyurea (H8627, 8 mM), blebbistatin (B0560, 50 µM), GM6001 (364206, 20 µM), 2-APB (Tocris Bioscience, 1224, 100 µM), BAPTA AM (Invitrogen, B6769, 25 µM), pyrrophenone (Cayman Chemical, 13294, 0.1 µM), importazole (SML0341, 20 or 40 µM), doxycycline (D3072, 10 nM), paraformaldehyde solution (Electron Microscopy Sciences, 1517-S), Triton X-100 (T9284), Tween-20 (P7949), Nonidet P-40 substitute (VWR, M158), Everyblot blocking buffer (BioRad, 12010020), TBS (Quality Biological, 351-086-101) and bovine serum albumin (A7030).
Photolithography and device fabrication
PDMS microfluidic devices, consisting of an array of parallel channels with a fixed channel length of 200 µm and different heights and widths were fabricated as described previously52,53,54. On the basis of their cross-sectional areas, channels were classified as moderately confining (W × H = 10 × 10 µm2), confining (W × H = 10 × 3 µm2) or tightly confining (W × H = 3 × 3 µm2). For cell migration experiments, channels were coated with 20 µg ml–1 of rat tail collagen I (Gibco, A1048301).
Microfluidic device seeding and live-cell imaging
Cell seeding was performed as described previously17. To create a chemotactic gradient, the bottom three wells of each device were filled with serum-free DMEM (1% penicillin/streptomycin), whereas the top well was filled with serum-containing DMEM (10% FBS and 1% penicillin/streptomycin).
Cell phenotype analysis
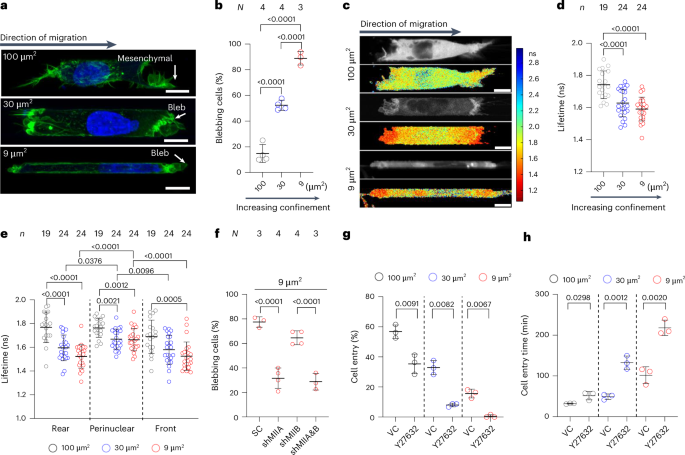
Cells were allowed to migrate in PDMS-based channels for 4–5 h at which timepoint, the cells were fixed and stained with Hoechst 33342 and phalloidin (as detailed below) and observed using a Nikon AXR confocal with ×40 water objective or an inverted Nikon Eclipse Ti microscope using a ×40 air objective. Migration phenotype was manually tabulated using the criteria described in refs. 11,17.
To calculate the percentage of cell entry, we counted the total number of cells within a distance of 50 µm from the channel entrances and quantified the fraction of these cells that fully entered the microchannels. Cell entry time was defined as the duration between cell protrusions first extending into the interior of the microchannels and full cell entry.
Effects of nuclear import and RhoA activity on cytoplasmic anillin level
HT-1080 cells were seeded into 96-well glass-bottomed plates (Cellvis, P96-1.5H-N) at 4,000 cells per well. After 24 h, cells were treated with either importazole for nuclear import inhibition or doxycycline for inducible overexpression of GFP-RhoA (Q63L). Following 24 h of treatment, cells were washed and fixed with warm 4% paraformaldehyde in phosphate-buffered saline, stained for endogenous anillin and imaged with an ImageXpress Micro Confocal high-content microscope (Molecular Devices) similar to the protocol described above.
Live-actin imaging
Cells harvested from flasks were resuspended in a medium containing 1:1,000 SPY650-FastAct (Cytoskeleton; CY-SC505, 1,000× stock in dimethyl sulfoxide), seeded into microfluidic devices, and kept for 3.5 h in an incubator before imaging.
Fluorescence imaging and quantification
All fluorescence data, except for those in Fig. 2d, Extended Data Fig. 2a and Fig. 5a, were acquired on a Nikon A1 or AXR confocal microscopes (Nikon) using a ×63 oil objective with a numerical aperture of 1.4 or ×40 water objective with a numerical aperture of 1.15. Also, 640-nm, 567-nm, 488-nm and 405-nm lasers were used for imaging. Fluorescence intensity was quantified using ImageJ (v.2.16.0/1.54p). For anillin, the front and rear plasma membranes and the nucleus were selected as depicted in Extended Data Fig. 2d. Fluorescence intensity was measured in selected regions and normalized to the total cell fluorescence intensity. In 3D collagen and viscoelastic alginate gels, the cell pole with the highest fluorescence signal was quantified and identified as the cell periphery.
For pMLC quantification, fluorescence intensities for the whole cell, nucleus, and front and rear plasma membranes were quantified. Cytoplasmic pMLC intensity is the mean fluorescence of the cytoplasmic area excluding the nucleus (Extended Data Fig. 2d). Mean fluorescence of the cell front and rear is normalized to the mean cytoplasmic intensity excluding the nucleus and the front and rear membranes.
ACE quantification
Cell z scans were taken at 0.5-µm intervals, and the z plane with the strongest membrane anillin signal was chosen. GFP-anillin intensity was analysed along a 4-pixel-wide linescan running between the membrane region displaying anillin signal and the cytoplasm. Cells on 2D, 3D and moderately confining channels are rendered positive for ACE if they have ≥5 µm of membrane region with anillin intensity at least twice that of the surrounding cytoplasmic region. Similarly, in confining channels, ACE-positive cells are considered those with at least twice the anillin intensity signal at the cell poles relative to the surrounding cytoplasmic region.
NE rupture imaging and quantification
Cells expressing NLS-mCherry were imaged on a Nikon A1 confocal microscope using a Plan Apo ×20 air objective with a numerical aperture of 0.75 and a resolution of 1,024 × 512 pixel2. A central z plane of cells inside a confining channel or z stacks at a 0.5-µm interval were acquired. For the quantification of NE ruptures, the reduction in nuclear NLS-mCherry signal with the corresponding increase in its cytoplasmic intensity is considered a rupture event. Conversely, the recovery of the nuclear signal accompanied by a reduction in the cytoplasmic signal is marked as an NE repair event. Cells were analysed from when their nuclei reached the channel entrances until the cell protrusions reached the channel exit. Cells whose nuclei were obstructed by particles or cellular debris during confined migration were excluded.
The videos of migrating cells in confinement were manually inspected to identify the the x and y positions of the centres of nuclei in the first frame corresponding to full nuclear entry into microchannels. The nuclei images were first segmented using a custom program developed in MATLAB_R2023b55,56,57. The area (A), perimeter (P), and the long-axis and short-axis lengths of the segmented nuclei were then computed using the image processing toolbox in MATLAB. The aspect ratio was calculated as the ratio of the long-axis length and the short-axis length, and the circularity is 4πA/P2.
Manual linescan analyses with ImageJ were used to identify video frames indicating NE rupture events as signalled by the abrupt increase in the cytoplasmic signal and decrease in the nuclear NLS-mCherry signal. In parallel, a 10-pixel-wide linescan with ImageJ in the GFP-anillin channel was used to identify video frames corresponding to the formation of front or rear ACEs. Time stamps of all changes in the nuclear/cytoplasmic NLS-mCherry and ACE detection in the GFP-anillin channel were recorded in Excel (v.16.97.2) sheets and used for the calculation of the frequency of NE ruptures and timing of the front and rear ACEs with respect to NE rupture. Similarly, linescans of the nuclear and diffuse perinuclear cytoplasmic GFP-anillin with ImageJ were used to detect the timing of nuclear GFP-anillin exit.
FLIM of RhoA FRET sensors
Confocal FLIM of live cells that were stably expressing the RhoA2G sensor was performed as described previously17 using a ZEISS LSM 780 microscope and a PicoQuant system consisting of the PicoHarp 300 time-correlated single-photon-counting module, two hybrid PMA-04 detectors and a Sepia II laser control module.
FLIM image processing, segmentation and quantification
The FLIM data were processed as described previously17 using SymPhoTime 64 (PicoQuant) software. Pseudocolour heat-map images showing the fluorescent lifetimes within predefined ranges were prepared with a customized ShowFluorLifeData MATLAB R2023a script55.
HEMICA device preparation and seeding
An array of parallel channels with a fixed channel length of 200 µm and different heights and widths were designed via soft photolithography, as described previously54,58. HEMICA microchannels with elastic moduli of 8 kPa and 21 kPa were fabricated, and coated with collagen type I, as previously described31.
Alginate gel preparation, fabrication and mechanical testing
High-molecular-weight I1G alginate (~260 kDa) was purchased from KIMICA and was irradiated by a cobalt-60 source to produce low-molecular-weight alginate (~27 kDa). RGD-coupled alginate was prepared by coupling the peptide GGGGRGDSP (Peptide 2.0) using carbodiimide chemistry. The alginate was then purified by dialysis (3,500 molecular-weight cut-off) against deionized water containing sodium chloride for 3 days, treated with activated charcoal, sterile filtered, lyophilized and reconstituted in DMEM (1% penicillin/streptomycin, no FBS) following previously reported methods34. Calcium sulfate (CaSO4) was mixed with alginate as a source for the release of crosslinking calcium ions. The mixture was transferred to a glass plate coated with Sigmacote, covered and allowed to gel for 45 min. Gel discs, which were 15 mm in diameter and 2 mm thick, were equilibrated in DMEM (1% penicillin/streptomycin, no FBS) for 24 h before mechanical testing. The elastic modulus and stress relaxation properties of alginate hydrogels were measured by compression tests of the gel discs using an MTS Criterion Series 40 Tensile Tester. The gel discs were compressed to 15% strain at a deformation rate of 2 mm min–1 and a 100-Hz data acquisition rate. For the relaxation process, the compression strain was kept at 15%, as the load was recorded over time. The elastic modulus was derived from the slope of the linear region of the stress–strain curve (~5–10% of strain). Stress relaxation properties were quantified by relaxation half-time (t1/2), which is the time for the initial stress to be relaxed to half its value during stress relaxation test.
Tumour implantation and intravital multiphoton microscopy
Athymic nu/nu female mice were obtained from the Department of Experimental Radiation Oncology, M.D. Anderson Cancer Center. Dorsal skin-fold chambers were mounted on 8-to-12-week-old female athymic nu/nu mice as described previously35. In brief, the skin-fold chamber was mounted on a skin flap on the back to cover the deep dermis after surgically removing the opposite side of the skin. One day post-surgery, pelleted HT-1080 cells (2.5–5 × 105 cells in 2–4 µl) stably expressing NLS-mCherry and GFP-anillin were injected into the dermis with a 30-G needle. Three tumours per chamber were implanted and monitored for up to 11 days. Intravital microscopy was performed on a LaVision TrimScope II scanner with three titanium–sapphire lasers (Chameleon-XR, Coherent) and two optical parametric oscillators compact systems (APE/Coherent; tunable excitation wavelengths range between 800 nm and 1,300 nm) on days 2–11 to monitor the tumour growth and subcellular distribution of NLS-mCherry and GFP-anillin. Next, mice were anaesthetized with isoflurane (1–3% in oxygen), placed on a temperature-controlled stage (37 °C) and the chamber was mounted on a holder. Blood vessels were visualized by a retro-orbital injection of 70-kDa dextran (Invitrogen/Thermo Fisher) labelled with Alexa Fluor 750 (1 mg per mouse). Imaging was performed using an Olympus XLPLN25XWMP2 ×25 water objective (numerical aperture, 1.05; working distance, 2 mm). Sequential 3D stacks were acquired with three excitation wavelengths (880 nm, 1,090 nm and 1,280 nm) in two consecutive scans. Emission was detected using the following band-pass filters: third-harmonic generation (1,280 nm; ET450/60 nm), mCherry (1,090 nm; ET595/40 nm), SHG (1,090 nm; ET525/50 nm), Alexa Fluor 750 (1,280 nm; ET810/90 nm) and GFP (880 nm; ET525/50 nm; Chroma Technology). The 3D volumes were acquired for up to a 250-µm penetration depth at a step size of 5 µm. Time-lapse recording with a frame interval of 20 min was performed for a maximum duration of 5 h. The 3D image stacks were reconstructed as the maximum-intensity z projection, stitched and analysed using NIH ImageJ.
In vivo image analysis
To quantify the NE rupture events in vivo, the mean grey values of the the N/C ratio for the NLS-mCherry channel were analysed from 2–3 z-projection image slices of the time-lapse sequences and plotted as the signal intensity profiles over time. NLS positivity in the cytoplasm was determined by an average N/C ratio of ≤2.5. The redistribution of anillin to the plasma membrane was quantified as the ratio between the membrane and cytoplasmic (M/C) mean grey values for GFP-anillin. Regions of interest were defined by manual segmentation along the inside of the nucleus and cytoplasm or along the cell edge. Anillin positivity at the membrane was defined as having at least 5 µm of membrane region with the anillin intensity averaging over time at least twice the intensity of the surrounding cytoplasmic region.
To correlate the percentage of cells with cytoplasmic anillin with the geometry of the local invasion environment, the constriction width was quantified using combined 3D SHG, fluorescent dextran and cell-based fluorescence in the orthogonal direction from the invasion path. Only cells bordered by a detectable SHG signal above the background were analysed. We associated these data with the number of cells displaying ACEs in each invasion zone, with ACE quantification performed as described in the relevant section. This analysis was validated through an independent, blinded review.
Ex ovo chick embryo cancer xenograft model
Fertilized White Leghorn chicken eggs, acquired from the University of Alberta Poultry Research Centre, were maintained in a humidified incubator at 38 °C. After 4 days of incubation, the embryos were removed from their shells and maintained under shell-less conditions in a covered dish at 38 °C and 60% humidity, as previously described46,47,48.
For the primary tumour or primary tumour invasive front imaging, day-10 chicken embryos were injected with 1 × 105 mCherry-labelled HT-1080 cells or mCherry-labelled HT-1080 cells expressing either WT GFP-anillin and HA-Ect2 (ANLN/Ect2 (WT)) or anillin and Ect2 dual mutant (ANLN-Δ3/Ect2-DHmut) in phosphate-buffered saline directly in between the CAM ectoderm and endoderm layers. Sterilized, circular coverslips (22 mm in diameter) were positioned on top of the tumour 1 day post-tumour cell inoculation, and image acquisition was performed 4–5 days later46,47,48.
For metastatic colony imaging, day-10 chicken embryos were injected intravenously with 2.5 × 104 mCherry-labelled HT-1080 cells or mCherry-labelled HT-1080 cells expressing either ANLN/Ect2 (WT) or ANLN-Δ3/Ect2-DHmut. Sterilized, circular coverslips (22 mm in diameter) were positioned on the CAM surface above metastatic colonies 1 day post-tumour cell injection. Metastatic colonies were allowed to develop for four more days, and single metastatic colonies were selected for visualization and analysis46,47,48.
In cancer cell extravasation experiments, 5 × 104 mCherry-labelled HT-1080 cells expressing either ANLN/Ect2 (WT) or ANLN-Δ3/Ect2-DHmut were injected into the CAM vasculature. Cancer cell extravasation was analysed 8 h post-injection, as described in ref. 59.
Image acquisition and analysis
The real-time imaging of cancer cell invasion was performed by acquiring a four-dimensional image series of single cancer cells within the CAM tissue, as previously described46,47,48, using a Nikon A1r upright microscope fitted with a temperature-regulated enclosure and a range of Nikon microscope objectives (×10, ×25 (water immersion) and ×63 (oil)). For the time-lapse analysis, time 0 was defined as the time of the first image capture. Here 20–50 individual cells were tracked (for each cell line used in the experiments) using a built-in object-tracking module in Volocity. The track velocity was calculated as the average speed of the track. The track displacement rate (productivity) was calculated using a built-in Volocity module as the total track displacement (straight-line distance from the first track position to the last) divided by the track time. Cancer cells were identified as anillin cytoplasmic high if the ratio of average anillin signal intensity (GFP) within the nucleus to the average anillin signal intensity within the cytoplasm was ≤3; the cells were identified as cytoplasmic low if the GFP-anillin nuclear/cytoplasmic ratio was ≥5. To quantify the cancer cell number per colony, ×25 z-stack images were acquired (2–5-µm step), and the cancer cells were manually counted using Nikon Elements software (v.5.21.00). For the quantification of invasive cancer cells at the primary tumour periphery, individual (×25) z-stack images were analysed using the Nikon Elements software. For anillin-GFP signal intensity quantifications, the primary tumour images (main tumour mass and invasive zone) were acquired at ×60 magnification. All the experimental data were plotted and analysed for statistical significance using the Prism analysis module.
Also, ×63 confocal images of the main tumour mass and invasive zones were obtained to quantify the anillin fluorescence intensity ratios. ImageJ was used to quantify the anillin or mCherry fluorescence intensity within the nucleus or cytoplasm within single optical slices (1 µm). For time-lapse tracking analysis, image drift was corrected using the ImageJ Stack_Reg plugin (Biomedical Imaging Group; http://bigwww.epfl.ch/thevenaz/stackreg/).
ACE quantification was performed using ImageJ. For cells inside the primary tumour masses, a 256 × 256-pixel2 region of interest was placed in the middle of the tumour mass. All the cells with at least half their nuclei inside the region of interest were included in the analysis. All cells that were found in the invasive zones were analysed. ACE quantification was performed as described in the relevant section. Cells that had no mCherry signal or whose NE was not distinguishable were excluded from our analysis.
Supplementary Methods contain additional information on the following experimental procedures: cloning, lentivirus preparation, transduction, and transfection; actin staining, immunofluorescence imaging and quantification; cell-cycle synchronization; flow cytometry analysis; western blotting; coimmunoprecipitation assays; and collagen- and alginate-gel-related assays.
Statistics and reproducibility
Data represent the mean ± standard deviation (s.d.) or median with 95% confidence interval from N independent experiments. All the experiments were performed in triplicate (three biological replicates), unless otherwise specified. The D’Agostino–Pearson or Shapiro–Wilk omnibus normality test was used to determine whether data are normally or log-normally distributed. Datasets with Gaussian distributions were compared using a two-tailed Student’s t-test, a one-way analysis of variance (ANOVA) test followed by a Tukey’s test for multiple comparisons or a two-way ANOVA test followed by a Sidak’s test for multiple comparisons. A Wilcoxon matched-pairs signed rank test was used to determine the statistical significance. Log-normal data were transformed using the Y = log[Y] formula before comparison. For non-Gaussian distributions, a non-parametric two-tailed Mann–Whitney test was used for comparing two conditions, whereas more than two groups were compared by a Kruskal–Wallis test followed by Dunn’s multiple comparisons. Analysis was performed using GraphPad Prism (v. 6, 7, 8, 9 or 10) software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.