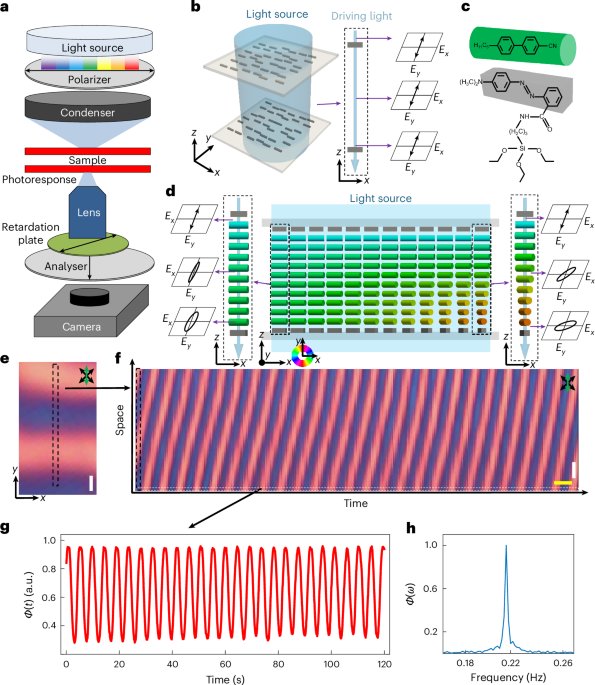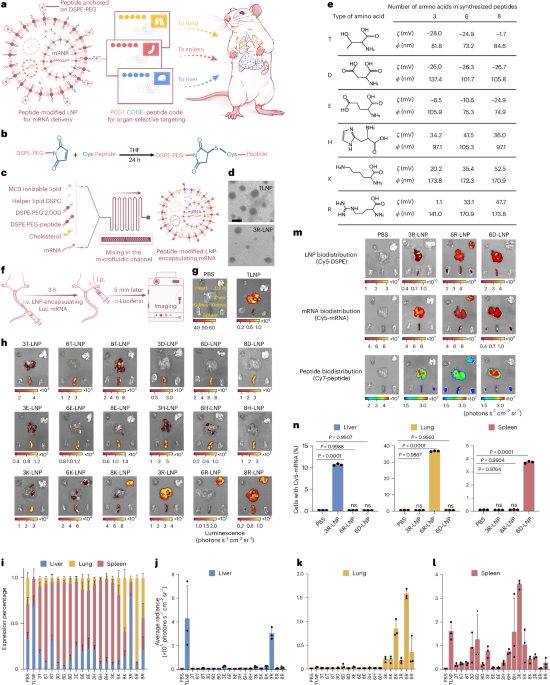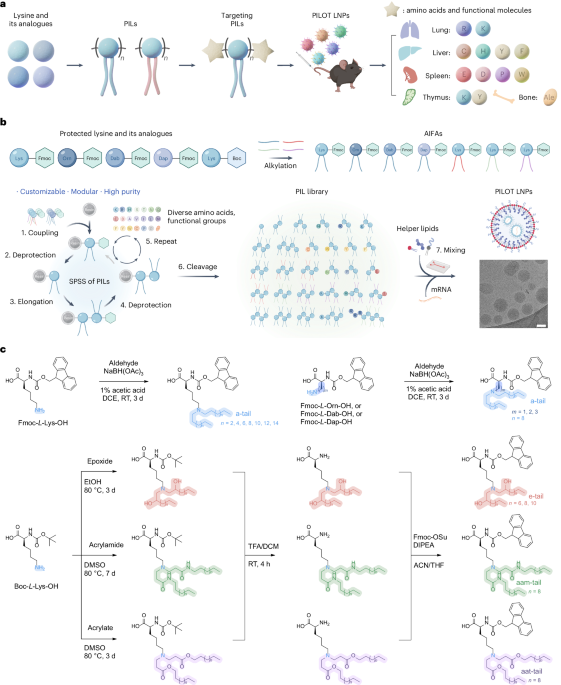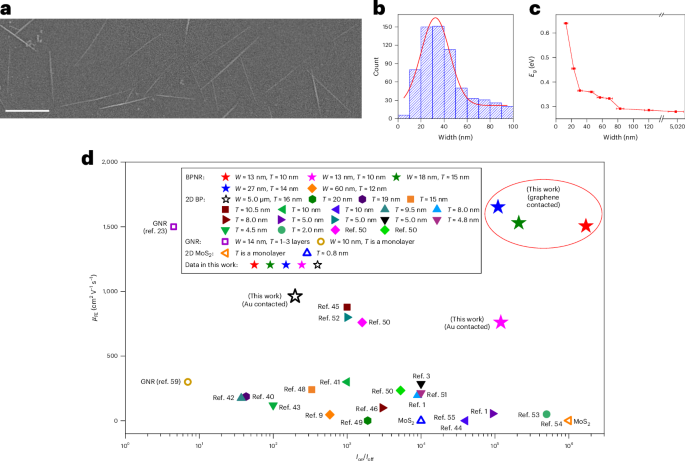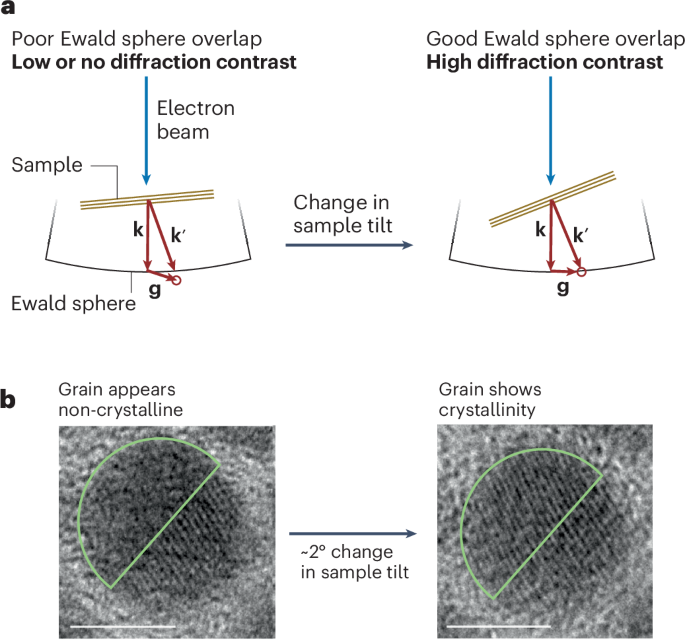
Directionality and dispersibility in peptide self-assembly
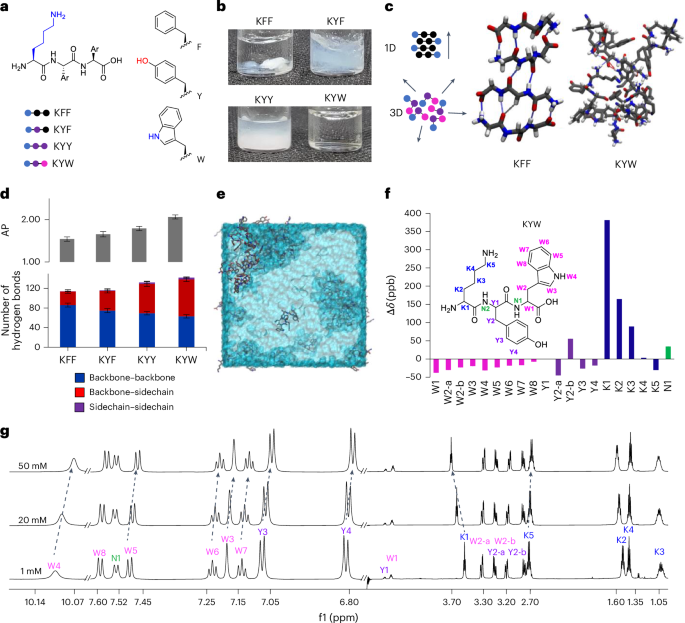
Tripeptide assembly is favoured by sequences of two aromatic amino acids complemented with a charged residue23,33. Aromatic amino acids (Y and W; Fig. 1a,c) add a diversity of interactions beyond backbone hydrogen-bond directionality, including side-chain π-stacking, hydrogen-bonding and dipole–dipole interactions, importantly, including those with water34. Lysine–phenylalanine–phenylalanine (KFF), KYF and lysine–tyrosine–tyrosine (KYY) undergo gelation and phase separation, but lysine–tyrosine–tryptophan (KYW) remarkably retains the appearance of a clear solution up to 20 mM in phosphate buffer23 (Fig. 1b). Notably, KYW assembly is strongly dependent on the ionic context and on forming fibres and gels in unbuffered conditions23,35.
We used atomistic molecular dynamics (MD) simulations to understand differences in assembly and hydrogen-bond interactions in KFF, KYF, KYY and KYW (Fig. 1d and Supplementary Fig. 11). First, exchanging F → Y → W leads to an increase in the aggregation propensity (AP; Fig. 1d and Supplementary Fig. 12). We analysed the contribution of the backbone–backbone (as a proxy for 1D self-assembly) and side-chain hydrogen bonds (Fig. 1d and Supplementary Figs. 13–16). Consistent with the increased AP, we observed a concomitant increase in the total number of hydrogen bonds in F → Y → W variants, with decreased backbone and increased side-chain hydrogen bonds resulting in non-directional assembly for KYW (Fig. 1d).
We investigated the concentration-dependent behaviour of the KYW peptide using extensive 1H nuclear magnetic resonance (NMR) analyses at 1, 20 and 50 mM in pH 7.5 phosphate buffer (Fig. 1f–g and Supplementary Fig. 17 and accompanying text). Across this range, KYW showed no line broadening or signal loss, indicating that it remains highly dynamic and fully soluble even at higher concentrations. The assembly process is driven primarily by π-type hydrophobic interactions, side-chain hydrogen bonding and electrostatic interactions, consistent with a dynamic, non-aggregating state.
The relative contributions of side chains and backbone interactions when substituting F → Y → W are also evident in the circular dichroism spectra (Supplementary Figs. 18–20). At a concentration of 5 mM, which is just above the critical aggregation for KYF and KYY (Supplementary Fig. 20a,b), the spectra show a carbonyl π → π* transition peak at 203 nm (Supplementary Figs. 18, 19a,b and 20a,b), suggesting backbone–backbone interactions, while the transitions of aromatic side chains may also contribute22. For KYW, this transition is less intense, while the appearance of the 229 nm positive signal that increases with concentration (Supplementary Figs. 18 and 19c) confirms the dominating influence of tryptophan side-chain interactions8,36.
Sequence dependence in tripeptide dispersions
We previously demonstrated differential assembly of tripeptide sequence isomers driven by side-chain-induced conformation selection33. We speculated that in a system stabilized by a multitude of possible side-chain and water interactions, these would not be defined by one dominant conformation but instead represented by a rich ensemble of conformers. We studied the six K/Y/W sequence isomers to determine the sequence-dependent self-assembly (Fig. 2a and Supplementary Figs. 21–26) and found lower variation in AP values (Fig. 2a and Supplementary Fig. 21) and similar ratios of side-chain/backbone interactions (Fig. 2a and Supplementary Figs. 22–25), consistent with the formation of non-directional ensembles. We also analysed the solvent exposure of tryptophan, measured through the solvent-accessible surface area (SASA; Fig. 2a and Supplementary Fig. 26) of the indole side chain. This was found to be sequence dependent, with tryptophan–tyrosine–lysine (WYK) and tryptophan–lysine–tyrosine (WKY) being the most contrasting in terms of AP and tryptophan solvent exposure (Fig. 2b).

a, From top to bottom, the AP score, tryptophan SASA and hydrogen-bonding interaction distribution analyses for K/Y/W sequence isomer MD trajectories. Averages and s.d. error bars were measured from three replicate simulations using 250 data points (last 50 ns) of each analysis set. Below is the ThT fluorescence emission intensity (50 µM) in the presence of peptide at 20 mM. Averages and s.d. error bars from three replicates. Data represent mean ± s.d. (n = 3). b, Snapshots for WKY and WYK showing non-directional self-assembly resulting in differential tryptophan environments and water binding (shown in blue) with zoomed-in areas showing a dry (top) and water-bound (bottom) tryptophan. c, The 3D fluorescence spectra showing the differential polarity and water interactions of tryptophan residues in solution. λem, emission wavelength. d, Chemical shift perturbation (Δδ in parts per billion) for WKY and WYK between concentrations of 50 mM and 1 mM. Each plot corresponds to a different sequence, with downfield shifts represented as positive values and upfield shifts as negative values. Each bar represents the average of three independent experiments, with s.d. error bars from three replicates. e, Concentration-dependent chemical shifts of the indole NH proton in K/Y/W sequence isomers, highlighting variations in the chemical environment and intermolecular interactions as concentration increases (1, 20 and 50 mM). Averages and s.d. error bars from triplicates.
Source data
Tryptophan exposure in peptide ensembles could be experimentally verified by measuring the tryptophan emission, which is widely used as a probe to detect the tryptophans’ environment and solvent exposure in proteins, with redshifted emissions indicating an increase in local polarity and blueshifts indicating hydrophobic environments37,38,39. These ensembles (20 mM) did not show evidence of phase separation when observed by optical microscope (shown for KYW in Fig. 1b). We measured the emission spectra of all sequence isomers in soluble dispersions (Fig. 2c) and found that emission maxima (excitation wavelength λex = 280 nm) range from 357 nm to 370 nm, with the most redshifted emission corresponding to the most hydrated tryptophan (WYK) and the most blueshifted emission corresponding to the most hydrophobic tryptophan environment (WKY; Fig. 2b). The 3D excitation/emission spectra of contrasting sequences (Fig. 2c) differed by the appearance of a new band in WYK’s spectrum at λex = 318 nm, which is also prominent in tyrosine–lysine–tryptophan (YKW) and tyrosine–tryptophan–lysine (YWK). We also found that the intensity and absence/presence of the band is concentration dependent, suggesting its origin from an emissive state of a supramolecular arrangement (Supplementary Fig. 27).
Concentration-dependent NMR studies on all K/Y/W sequence isomers revealed that while all maintain high dynamicity and solubility up to 50 mM, π-type, cation–π, hydrogen-bonding and electrostatic interactions contribute across all isomers, and the nature and strength of intermolecular interactions vary with sequence (Fig. 2d, Extended Data Fig. 1 and Supplementary Fig. 28 and accompanying text).
The local chemical environment of tryptophan residues was probed by following the NH (W4) and CH (W3) indole protons in the 1H NMR spectra of the six K/Y/W peptide solutions at variable concentrations and showed contrasting concentration-dependent chemical shifts of tryptophan protons (Fig. 2e and Supplementary Fig. 29). The indole-ring protons in WYK shift downfield as the concentration increases, indicating their positioning in an electron-poor environment and the formation of a polar network and hydrogen bonding, while (minimal) upfield shifts were observed for WKY, in line with the formation of π-stacking-driven aggregation. The other four sequences exhibited moderate upfield shifts, less pronounced than those of WKY, in agreement with the MD simulation data (Fig. 2a).
To quantify the degree of assembly across peptide sequences, we monitored changes in thioflavin T (ThT) emission upon co-incubation with 20 mM peptide solutions (Fig. 2a), revealing enhanced fluorescence consistent with the formation of dynamic hydrophobic pockets. This sequence-dependent ThT response, aligned with calculated AP scores and supported by NMR chemical shifts, confirms the presence of soluble dispersions featuring dynamic hydrophobic domains.
To further probe dynamic ensembles in solution, we conducted fluorescence spectroscopy across increasing concentrations (2–20 mM), observing progressive tryptophan quenching (Supplementary Fig. 30). Concentration-dependent diffusion changes, analysed by 1H diffusion-ordered spectroscopy (DOSY) NMR, were performed at 1, 20 and 50 mM (Supplementary Fig. 31), revealing decreased diffusion with increasing concentration. The peptide-to-water diffusion ratio suggests small soluble aggregates, consistent with unbroadened 1H NMR signals. The 1H–1H nuclear Overhauser effect spectroscopy (NOESY) NMR spectra for all six K/Y/W tripeptides highlight water interactions, predominantly at W4 and W3 (Supplementary Fig. 32). WYK and YWK exhibit enhanced water contacts at positions W2 and W1, consistent with higher SASA values (Fig. 2a), while WKY also shows a W1–water correlation that aligns with the 1H NMR downfield shift (Fig. 2d and Supplementary Fig. 28 and accompanying text), likely due to its N-terminal location.
Evaporation-driven assembly
Upon investigating the fluorescence of KYW peptide assemblies using confocal microscopy, we observed a remarkable phase separation behaviour triggered by localized evaporation during drying. Given the (competing) role of water in modulating interactions in these dynamic ensembles, in particular those involving tryptophan40, the observation led us to study the assembly during gradual removal of water through the evaporation of sessile droplets41,42,43 (Fig. 3a).

a, Representation of evaporation-driven self-assembly in sessile droplets. b, Evaporation patterns over time, with the differential assembly of KYW clearly visible. Scale bar, 1 mm. PBS, phosphate-buffered saline. c, Microscopy time course for evaporation-driven assembly of KYW. Scale bar, 50 µm. d, Bright-field microscopy images showing 1D assembly for KYY (left) and the formation of spherical particles upon drying for KYW (right). Scale bar, 10 µm. e, The dynamic droplet fusion process of WKY is recorded by time-lapse confocal microscopy. From 0 s to 14 s, a transient fusion of two liquid–liquid phase separation droplets is seen. By 430 s, the rigidification of the droplets and appearance of pores on the surface are seen. f,g, Time-lapse confocal analysis (f) revealed the onset of pore formation upon solidification. The moment that the droplet stationized was set as 0 s. The spectrum of bright-field images across the particle (white dashed lines) during the solidification process were profiled (g) for quantitative visualization.
Source data
Macroscopically, when observing tripeptide assembly in drying droplets starting from 20 mM peptide dispersions, we observed contrasting behaviours: a strongly scattering opaque ring upon drying for KYW, in contrast to KFF, KYF and KYY, which lead to heterogeneous semi-transparent structures (Fig. 3b and Supplementary Videos 1 and 2). When observed by microscopy (Fig. 3d and Supplementary Fig. 33), the differential morphologies (1D fibres versus 3D spherical objects) are evident (Supplemental Fig. 34 showing KFF and KYF). Over time, KYW and its five sequence isomers all display a remarkable drying-induced phase separation behaviour, giving rise to the formation of films composed of densely packed spherical particles (Fig. 3d, Supplementary Video 3 and Extended Data Fig. 2a,b), contrasting with KFF, KYF and KYY, which lead to 1D structures resembling previous observations for the evaporation-driven assembly of peptide derivatives43 (Supplementary Fig. 34, Extended Data Fig. 2a and Supplementary Videos 4–6). We propose that the multitude of possible interactions and the flexibility of the backbone enable the K/Y/W peptides to more effectively reconfigure and adapt to the newly formed interfaces that form upon water evaporation. The observed directionality (in the case of KFF, KYF, KYY) or lack of directionality (in the case of K/Y/W) initiates at the molecular level and clearly translates hierarchically to macroscopic scales. We observe complex drying patterns related to the solvent and solute gradients in the drying of droplets, with phase separation occurring at the drying front due to concentration gradients inside the drying droplet. Streaks observed during drying are a consequence of complex Marangoni flows in these drying droplets (Extended Data Fig. 2c–f and Supplementary Video 7).
Based on the previously reported interfacial supramolecular aggregation of tripeptides in oil–water emulsions35, we hypothesized that the ensembles would lead to interfacial aggregation and a reduction in surface tension. We measured the dynamic tension profile via the pendant drop method44 of the six K/Y/W tripeptides, which have contrasting tryptophan solvent exposures and environments. The dynamic surface tension indicates that the peptide sequence dictates the surface activity, with a general trend observed in which less solvent-exposed tryptophan (in WKY) gives rise to higher surface activity (Supplementary Fig. 35), demonstrating that the peptide ensembles dynamically interact at the air–water interface.
Time-dependent cryo-electron microscopy (cryo-EM) images illustrate the nanoscale structural transformations from amorphous aggregates to spherical droplets to porous droplets observed at the early stages of assembly (Extended Data Fig. 3). Dynamic droplet fusion and subsequent solidification is captured by time-lapse confocal microscopy (Fig. 3e, Supplementary Fig. 36 and Supplementary Video 8). Time-dependent confocal analysis revealed the onset of pore formation upon solidification (Fig. 3f,g). Time-dependent fluorescence spectroscopy during the drying-induced assembly gave rise to a decreased emission due to concentration-induced quenching, followed by a sharp fluorescence increase upon liquid–liquid phase separation (5–10 min), and subsequent quenching during droplet solidification (Supplementary Fig. 37).
Upon analysis of the tryptophan emission in the dried films at 5% relative humidity, we noted a general loss of emission from the 318 nm band in the 3D excitation/emission spectra, with spectra of the dried films now suggesting a redistribution of conformations (Supplementary Figs. 38 and 39). The original variable tryptophan exposure was partly recovered by temporarily exposing the solid film to high humidity (95% relative humidity). These data highlight the adaptive nature of the assemblies, in which tryptophan partitioning reversibly changes depending on the water competing via weak multivalent interactions.
Formation of buoyant droplets and porous particles
Upon microscopy analysis of the dried particulate films, we observed the expected spherical objects, but also porous, disc-shaped particles of varying sizes with flat surfaces (Fig. 4a). These structures are found at the top of the sample (Fig. 4b,c and Extended Data Fig. 4), suggesting that they are formed at the air–water interface. The observation suggests that, in contrast to typical dense coacervate droplets that form as a sediment, the structures studied here are low-density, buoyant condensates. The observation of flattened 2D hemispherical objects suggests that these particles are initially liquid, and due to their low density, they rise to the interface, where they deform, flatten and solidify. This optical observation (Supplementary Fig. 41) is confirmed using focused ion beam scanning electron microscopy (FIB-SEM; Fig. 4c), which shows a part-circular cross-section. The pore size distribution ranged from 30–500 nm (Supplementary Fig. 40).

a, Schematic representation of evaporation-driven assembly of buoyant liquid droplets that settle at the droplet interface and, upon drying, form half-dome-shaped particles. LLPS, liquid–liquid phase separation. b, SEM image showing a mixture of porous spheres and half-dome particles at the interface and also showing surface pores. c, FIB-SEM analysis confirming the half-dome shape and revealing the porous structure inside the dried particles. Note that the fine granular structures on the surface are from sputter-coated gold. d, TEM analysis of glutaraldehyde-crosslinked dissociated KWY particles reveals that non-surface particles are highly porous spheres. e, AFM analysis showing porous half-dome particles. f, The plot of Young’s modulus showing the stiffness of fibres or particles formed by the indicated peptides. A total of 15–36 particles or fibres were profiled for each sequence. In the box plots, the centre line shows the median, the box edges delineate the first and third quartiles and the whiskers show the range of values.
Source data
We propose that the observed buoyancy is related to the formation of gas bubbles inside liquid droplets (Fig. 4a). This may be a consequence of a loss of solvating water from the peptide structures, water that is replaced with air and leads to micro-cavitation, resulting in a porous structure upon dehydration (Fig. 4a). Notably, degassing of the peptide dispersion prior to evaporation-driven assembly leads to smaller particles of more spherical morphology (Fig. 5e). This observation indicates that the buoyant nature of the droplets relates to their gas content, and it can be regulated. Non-surface particles are smaller and spherical, indicating fusion and flattening at the air–water interface (Fig. 4b,c). Transmission electron microscopy (TEM) analysis of glutaraldehyde-crosslinked KWY particles reveals the porous and spherical structure of the non-surface particles (Fig. 4d). The hemispherical porous structure was further confirmed by atomic force microscopy (AFM; Fig. 4e and Extended Data Fig. 4). A Young’s modulus from Derjaguin-Muller-Toporov (DMT) model fitting analysis demonstrated that these particles, formed by all sequence isomers, are mechanically stiff (6 GPa) due to the efficient packing (Fig. 4f), in line with previously reported FF derivatives20,21.

a,b, Temperature dictates the size distribution of the dried peptide particles. KWY shows a reduction in particle size and more homogeneous size distribution as temperature increases during evaporation. Scale bar, 10 µm. A total of 368–1,456 particles at each temperature were analysed. c, The 20 mM WKY peptide was dissolved in Na/PB at concentrations of 25, 50, 100, 250, 500 and 1,000 mM. A bright-field image of dried WKY in 500 mM PB is shown. Scale bar, 50 µm. d, Plot of 296–662 Imaris-3D-rendered particles at each concentration demonstrates the reduction in particle size and increase in homogeneity with the increase of salt concentration. e–g, The peptide solution was degassed, followed by the evaporation assay; the example of WKY is shown (e). Imaris 3D rendering demonstrated a reduction in particle size and increase in sphericity, as plotted in f and g, respectively. Scale bar, 5 µm. h, Fully reversible formation of porous peptide particles upon addition of water and re-evaporation. Representative time-lapse images of KWY are shown. Scale bar, 50 µm. The macroscopic images of 5 µl peptide solution drops at each stage are shown in the insets. Scale bar in the insert, 500 µm. In the violin plots, the horizontal lines show the median as well as the 25th and 75th percentiles.
Source data
Size control, shape control and reversibility
We first investigated whether the size distribution of the particles formed could be controlled. Given the important role of water evaporation in this process, we reasoned that by simply increasing the temperature, evaporation would occur faster and consequently, the formation of solid particles would be accelerated. We observed that temperature increase leads to more homogeneous and smaller particles (Fig. 5a,b and Extended Data Fig. 5a,b). The combination of reduced buoyancy, due to lower gas content at elevated temperature, with the acceleration of the phase transition and solidification yields particles that are monodisperse (Extended Data Fig. 5c,d and Supplementary Fig. 41). Another approach to control sphericity was through an increase of the buffer concentration, which results in smaller and monodisperse particles (Fig. 5c), observed through an accelerated phase change and solidification (Fig. 5d and Extended Data Fig. 5e).
As an alternative method to control particle size, we removed the air from the solution through sonication under a vacuum, followed by storage under argon gas, and examined the drying process in a degassed solution under argon. Live confocal time-lapse imaging of the particle-forming process revealed smaller droplets (Extended Data Fig. 5f). Further Imaris 3D reconstruction and analysis demonstrated a 20-fold reduction in particle volume and increase in sphericity in the degassed samples compared with a prevalence of disc-shaped particles in non-degassed samples (Fig. 5e–g and Supplementary Videos 9 and 10). Consistent with the role of air in pore formation, degassed peptide particles show fewer and smaller pores (Extended Data Fig. 5g).
Next we probed the reversibility of the process. Upon a re-introduction of a 5 µl droplet of water on top of a dried film, the structures immediately redispersed, as shown in Fig. 5h. When this droplet was left to dry again, the particle film reappeared after a few minutes, demonstrating that the process is fully reversible (Supplementary Video 11). These observations strongly contrast with those previously reported for peptide materials, where the formations of 1D and 2D structures are highly stable due to a large enthalpic gain from cooperative hydrogen bonds, which are not readily reversed20,21. Peptide ensembles with multivalent, weak and dynamic interactions offer more reversible assemblies, as water solvation can compete with the energetics of the interaction space5.
Drying-induced encapsulation and protection
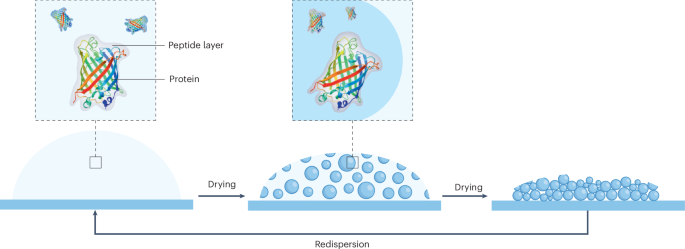
We propose that the spontaneous formation of tightly packed particles upon air-drying could be useful in encapsulation, protection and drying strategies, inspired by phase-separation protection mechanisms seen in biological systems45 (Fig. 6a). We first used the small organic fluorophore Alexa 488, which has been previously demonstrated to non-covalently conjugate through the sulfonate group and lysine side chains46. Upon introduction of the dye, it is encapsulated during KWY droplet formation and solidification (Fig. 6b and Supplementary Video 12). Analysis of the fluorescence intensity of the preload mixture of peptides and dye, the formed liquid droplets and the post-load solution revealed a highly efficient enrichment of the dye inside the particles. Similar efficient encapsulation was obtained for crystal violet, a positively charged hydrophobic dye (Extended Data Fig. 6).

a, Proposed mechanism for payload uptake involving dynamic complexation of payload molecules through dynamic side-chain interactions, followed by the molecules being surrounded to form droplets upon liquid–liquid phase separation. b, Confocal live imaging (left) of KWY peptide particles incorporating Alexa 488 shows efficient enrichment of the dye in peptide droplets. Scale bar, 20 µm. Fluorescence intensity analysis (right) of the preload solution (i), the droplet (ii) and the post-load solution (iii) at the frontier of emulsification. A total of 20 regions of interest of each group from six time-lapse frames were quantified, and the average intensity of the preload solution was set as 100 arbitrarily. Data represent mean ± s.d. (n = 20). Error bars represent s.d. c, Encapsulation assay of the EGFP protein by the WKY peptide was performed as described in b. Data represent mean ± s.d. (n = 20). d, Confocal and bright-field imaging of dried peptide particles reveal that the green fluorescence signals of EGFP remain in WKY peptide particles under ambient conditions for five days after solidification. e,f, Experiment to test the storage and redispersion ability of WKY with a schematic representation (e) of the experiment and the fluorescence emission at 510 nm (λex = 488 nm) of the corresponding solution (EGFP, EGFP + KYF and EGFP + WKY) over time (f). Data represent mean ± s.d. (n = 3). Error bars represent s.d. from three replicate experiments. g, The amount of reactive lysozyme in the dried particles with PBS or WKY at day 0 and day 15 under ambient conditions were measured by enzyme-linked immunosorbent assay (ELISA). Data represent mean ± s.d. (n = 3).
Source data
Next, inspired by the ability of dynamic interacting block copolymers with random distributions of side-chain functionalities to stabilize proteins through an adaptive matching of side-chain interactions to complementary patches on the protein surface31, we investigated the ability to encapsulate and stabilize enhanced green fluorescent protein (EGFP). We observed efficient encapsulation in WKY (Fig. 6c, Supplementary Fig. 42a,b and Supplementary Video 13). By comparing the encapsulation efficiency across the six sequence isomers, we conclude that both the Alexa 488 and EGFP encapsulation efficiencies are comparable for all peptide sequence isomers studied (Extended Data Fig. 7).
As EGFP fluorescence requires a folded conformation, we evaluated the preservation of structure in the dried state (Fig. 6d). After drying and subsequent rehydrating, the EGFP solution loses its fluorescence, whereas the WKY + EGFP sample shows retention of fluorescence emission after drying and storage in dried form for five days, followed by instantaneous re-dissolution (Fig. 6e,f). The control KYF + EGFP (fibrous assembly)23 reveals a decrease in the fluorescence (Fig. 6e,f and Supplementary Fig. 42c), suggesting a level of stabilization through protein–peptide interactions.
These results highlight that the drying peptide ensembles provide an environment where proteins are stabilized, including in the dried state, possibly through mechanisms analogous to dynamic interfacial complexation via the self-organization of side chains with a protein surface45 (Fig. 6a). Using either a desiccator or lyophilizer, we found comparable EGFP fluorescence recovery, indicating that further dehydration does not impair dry-state protein preservation (Supplementary Fig. 43). Under degassed conditions, we observed that the encapsulation of EGFP inside the particles was comparable to that at ambient conditions and does not depend on the microporosity (Supplementary Fig. 44).
The encapsulation mechanism in this system is distinct from conventional partitioning into preformed condensates. Here, small molecules or proteins first interact with dynamic peptide ensembles in solution through non-covalent side-chain interactions—particularly electrostatic and π-related contacts involving the lysine, tyrosine and tryptophan residues. These interactions result in complexation with the payload in solution, before phase separation resembling previously described dynamic surface-stabilization approaches32, enabling efficient engulfment of the payload during droplet formation.
A series of experiments support this mechanism: tryptophan fluorescence quenching confirmed the binding, before the liquid–liquid phase separation, of both positively and negatively charged aromatic dyes (crystal violet and Alexa 488; Extended Data Fig. 6a,b), and confocal microscopy demonstrated efficient encapsulation regardless of charge (Extended Data Fig. 6c–e). GFP variants with varied net charge47,48 revealed that a higher charge enhances encapsulation (Extended Data Fig. 8a–d), reinforcing the role of electrostatics and π-interactions. Furthermore, simultaneous encapsulation of multiple fluorescent proteins of differing charge and colour49 preserved the fluorescence over time, highlighting the system versatility (Extended Data Fig. 8e,f). Finally, functional lysozyme encapsulation and the retention of active enzyme after 15 days in the dried state confirmed both the stability and bioactivity preservation (Fig. 6g and Supplementary Fig. 45). The drying-induced assembly process was evaluated for potential scalability by heating, vacuum (Extended Data Fig. 9a) or producing small droplets for rapid drying, resulting in comparable particle formation and EGFP encapsulation within these sprayed micro-droplets (Extended Data Fig. 9b–d and Supplementary Video 14). These data suggest that K/Y/W isomer tripeptides offer potential for encapsulation, drying, storage and the redispersion of biomolecules.