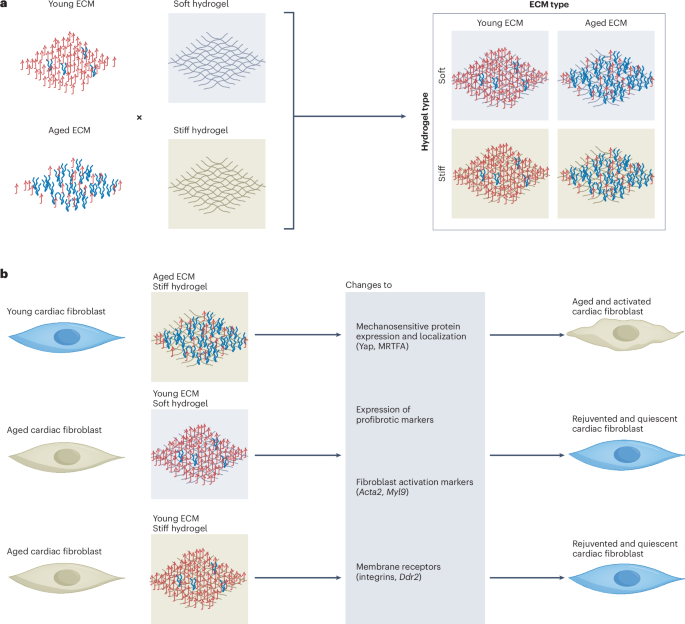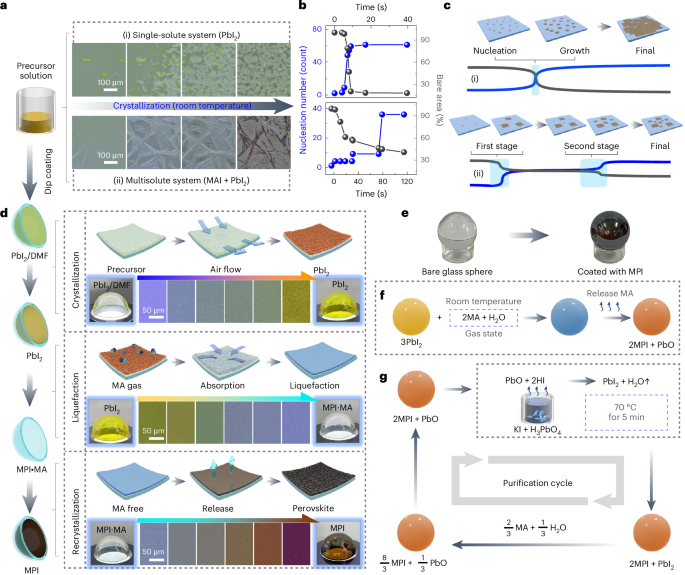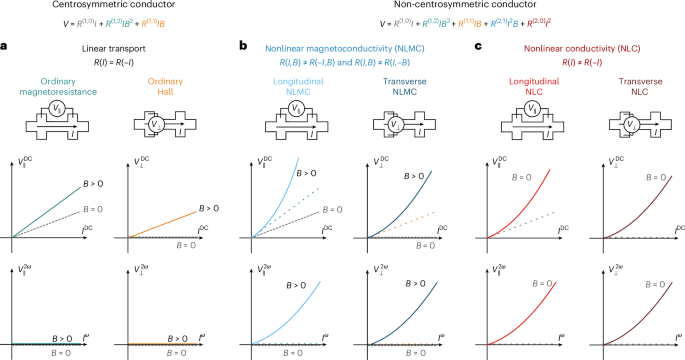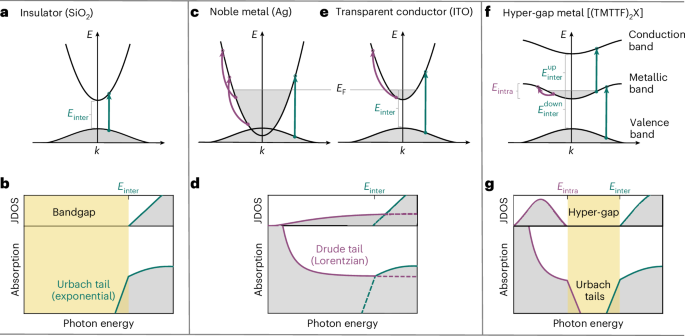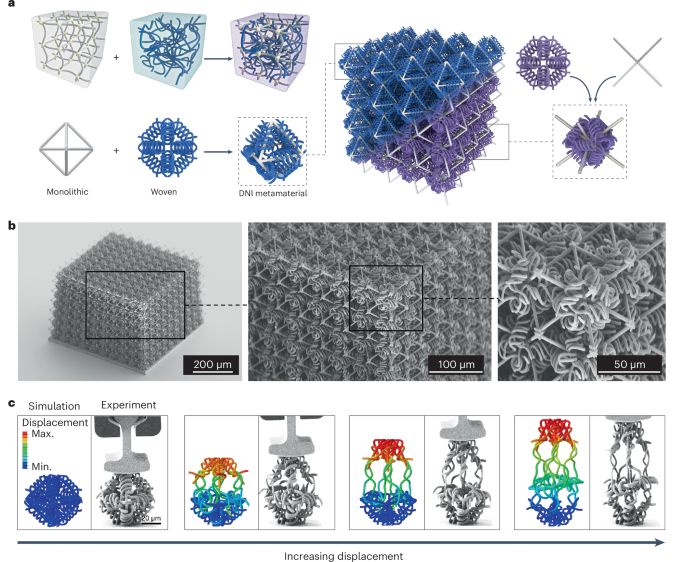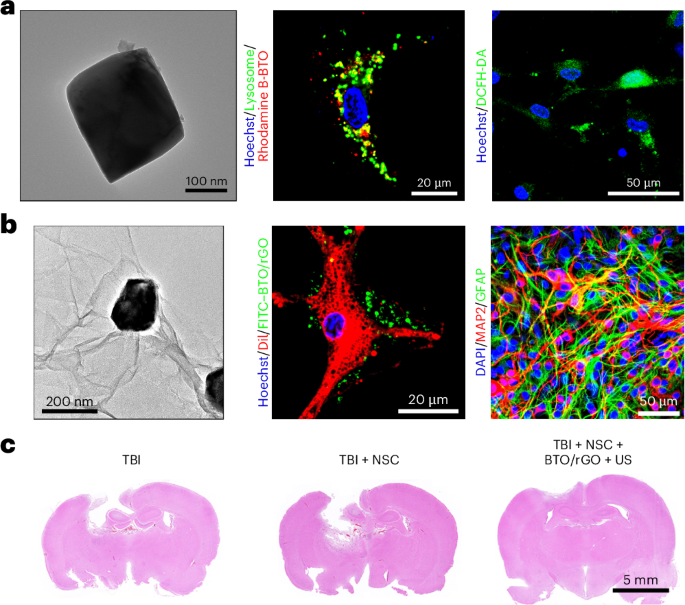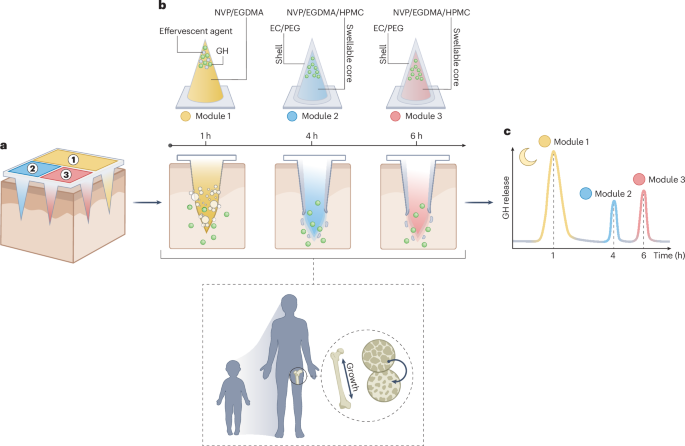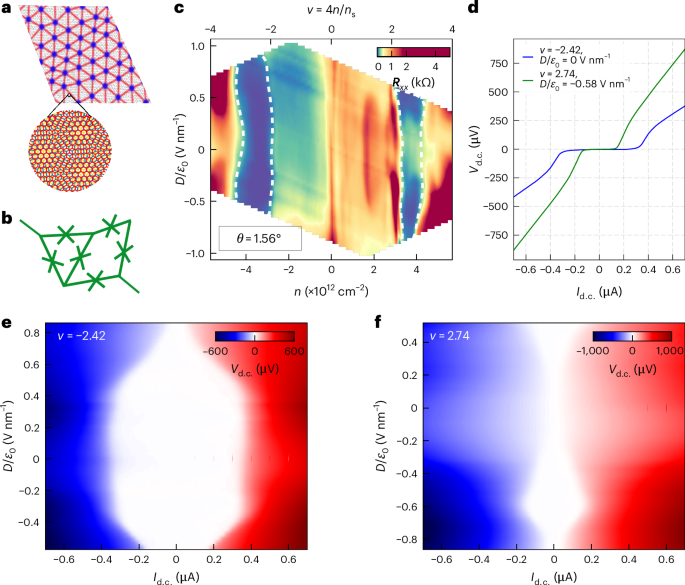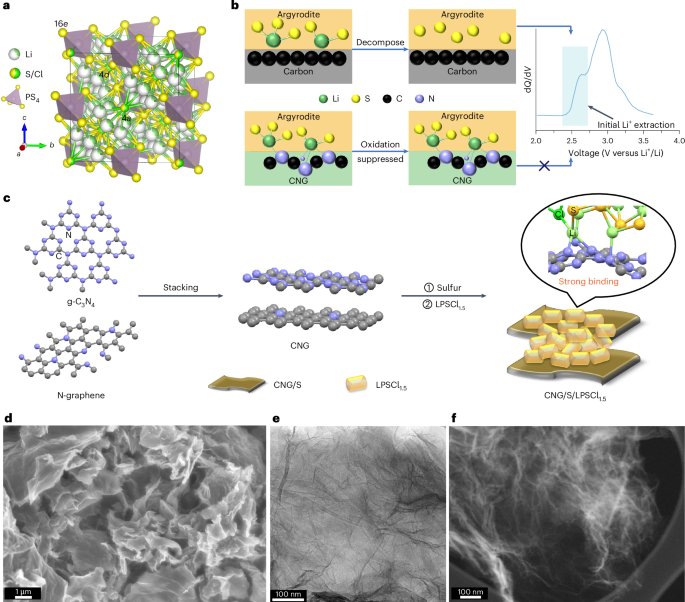
Preparation of coverslips and hydrogel solutions
Coverslips (ϕ = 15 mm) were cleaned via sonication during progressive washes with acetone, ethanol and MilliQ water for 10 min each and then air dried. For hydrogel attachment, coverslips were treated with a UV/ozone cleaner (Bioforce ProCleaner Plus) for 5 min followed by immersion in a solution containing 20 ml of ethanol, 600 μl of 10% acetic acid and 200 μl of 3-trimethoxysilylpropylmethacrylate for 5 min. Coverslips were then washed twice in ethanol and air dried before placing onto the tissue slice and hydrogel solution. The PA-based hydrogels used in this study mimicking the young (~10 kPa) and aged (~40 kPa) cardiac tissue stiffness were labelled as ‘soft’ and ‘stiff’ for easy reference, with their composition detailed in Supplementary Table 1. Prepared solutions of acrylamide and bis-acrylamide were prereacted with formaldehyde at 4 °C for 3 h at a stoichiometric ratio of >10:1 to ensure no excess formaldehyde in the solution16. Before hydrogel formation, Irgacure 2959 (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, Sigma-Aldrich) was added for UV photoinitiation. For hydrogel visualization experiments, separate samples of 10-kPa solution were made with Nile Blue-tagged acrylamide (Polysciences no. 25395-100) at 1:1,000.
Tissue handling, slicing and stabilization
Young (1–2 months) and aged (18–24 months) hearts were extracted from C57BL/6J strain female mice (obtained via NUS Animal Tissue Sharing Programme under Institutional Animal Care and Use Committee, National University of Singapore) after euthanasia and quickly transferred to sterile 1× phosphate-buffered saline (PBS) to minimize blood clot formation. The major vascular tissues superiorly attached to the hearts were then carefully removed with sterile surgical instruments. Before slicing the heart using a vibratome (Leica VT 1200S), the heart was embedded in a 4% low-melting-point agarose (Invitrogen) solution in sterile 1× PBS, with the orientation adjusted such that coronal slices are obtained during slicing. Agarose was kept on ice until solidified, attached to the slicing stage with superglue and loaded onto the vibratome. Following the initial trimmings of the agarose cube, the slicing speed was set to 0.18 mm s–1 and 150-μm slice thickness.
After tissue sectioning, vibratome slices were carefully transferred to a hydrophobic glass slide (dichlorodimethylsilane-treated) in a humid condensation chamber. The agarose surrounding the cardiac tissue was gently removed, as well as any excess liquid on the cardiac slices, by wicking away at the edges with a Kimwipe. Then, 21 ± 2 μl of the hydrogel solutions were added to each 150-μm-thick cardiac slice (solution volume was adjusted based on the volume of the tissue to fill the coverslip area) and incubated at 4 °C for 60 min in the dark. Crosslinking was then initiated using the same parameters as the plain hydrogel samples (35 mW cm–2 for 3 min). The samples were then transferred to a 12-well plate and kept in sterile MilliQ water at 4 °C before decellularization.
Decellularization of PA-stabilized cardiac tissue sections
All solutions were aseptically prepared, and all the experiment areas were sterilized frequently with 70% ethanol to prevent microbial contamination. SDC (Sigma-Aldrich) was dissolved in MilliQ water at 5% (w/v). The PA-stabilized tissue samples were exchanged with 2 ml per well of SDC solution and kept on a gentle shaker at room temperature for 2.5 days with one exchange at 24 h. The samples were then washed with MilliQ water and 10,000 U penicillin/streptomycin (pen/strep, Gibco) for 2 h. Subsequently, 2 ml of 300 kU ml–1 DNase-I solution (Sigma-Aldrich; dissolved in 0.15-M NaCl supplemented with 5-mM CaCl2 and 1% pen/strep) was added to each sample after discarding the pen/strep and gently shaking for 3.5 days followed by a final MilliQ water wash for 2 h.
Mass spectrometry
Freshly harvested and sliced cardiac tissues from young (2 months) or aged (24 months) mice were decellularized using the same protocol as described in the previous section using SDC and DNase-I. Then, dECM samples were snap frozen using liquid nitrogen and shipped on dry ice to BGI Genomics for further extraction steps followed by tandem-mass-tag-based quantitative mass spectrometry. Samples were ultrasonicated in an extraction cocktail containing EDTA and SDS-lysate buffer (7-M urea, 2-M thiourea, 20-mM Tris-HCl, pH 8.0) for 5 min on ice and centrifuged at 25,000g for 15 min at 4 °C. The supernatant was incubated in dithiothreitol (10 mM) and incubated at 37 °C for 30 min, followed by iodoacetamide (55 mM) treatment for 45 min. Proteins were precipitated using cold acetone at –20 °C and collected by a centrifuge at 25,000g for 15 min at 4 °C. Protein pellets were resuspended in a lysis buffer without SDS-lysate and centrifuged again to remove any insoluble residues. Quality control was performed after each round of protein extraction by Bradford quantification and sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Then, 0.5-M tetraethylammonium bromide was used to collect the protein by centrifuging three rounds at 12,000g and 20 °C, followed by trypsin digestion. Subsequently, peptides were labelled with a tandem mass tag for 2 h at room temperature, fractionized using Shimadzu LC-20AB liquid-phase system and freeze dried. The dried peptide samples were then injected into a Thermo Scientific UltiMate 3000 UHPLC connected to a mass spectrometer (Thermo Scientific Q-Exactive HF X). The ion source voltage was set to 1.9 kV; the MS1 scanning range was 350–1,500m/z and the resolution was set to 60,000; the starting m/z of MS2 was fixed at 100 and the resolution was 15,000.
Reconstituted solubilized dECM substrates
Young (1–2 months) or aged (18–24 months) murine hearts were sliced and decellularized using the same protocol as described in the previous section with SDC and DNase-I. The dECM samples were then snap frozen in liquid nitrogen and lyophilized overnight. Lyophilized dECM was then pulverized and solubilized2. Briefly, 1 mg ml–1 pepsin (Roche) was dissolved in 0.1-M HCl and added to milled dECM at a 10:1 ratio (w/w, dECM:pepsin) followed by constant stirring at room temperature. When a homogeneous solution was obtained, 1-M NaOH was used to neutralize the supernatant after removing insoluble residues. A Pierce BCA Protein Assay Kit (Thermo Scientific) was then used to quantify the total ECM protein amount. Solubilized dECM was diluted to 1 mg ml–1 with 1× PBS, coated onto 15-mm coverslips and incubated for 30 min in a condensation chamber at 37 °C.
ECM quantitative compositional assays
Quantitative compositional analyses of samples were performed for DNA, sGAG and collagen content. The sample’s DNA was extracted using phenol–chloroform and kept at −20 °C until applying the PicoGreen dsDNA assay kit (Invitrogen). Total sGAGs of the samples were obtained via papain (Sigma-Aldrich) extraction followed by Blyscan sGAG assay (Biocolor). For collagen quantification, the samples were first homogenized and hydrolysed in concentrated HCl at 120 °C for 3 h before being quantified using a hydroxyproline assay kit (Sigma-Aldrich). A microplate reader (Promega GloMax) was used for all the assays. Duplicate technical replicates with N = 3 biological replicates were measured along with standard curves for quantification, according to the suppliers’ instructions.
IHC
IHC of the native tissue sections and DECIPHER samples was carried out for ECM components and cellular structures (antibodies and dilutions are detailed in Supplementary Table 2). Briefly, samples were fixed with 4% formaldehyde (Sigma-Aldrich) and rinsed with 1× PBS before blocking and permeabilizing with 2% bovine serum albumin (BSA; Sigma-Aldrich) in 0.2% Triton X-100 (Sigma-Aldrich) for 30 min. The samples were then incubated with primary antibodies in 2% BSA on a gentle shaker at room temperature for 3 days followed by three times of 1× PBS washing and incubation of secondary antibodies and CF Phalloidin (Biotium) staining for 1 h at room temperature in the dark. To label any denatured collagen, Cy3-conjugated collagen hybridizing peptide (R-CHP, 3Helix) was diluted to 5 μM and used to stain the samples after CNA35 labelling according to the product’s instructions64,65. The samples were further stained with Hoechst 33342 (Invitrogen) and mounted using Fluoromount-G (Invitrogen). For hydrogel visualization experiments using Nile Blue-tagged acrylamide, the samples were co-stained with CNA35.
Immunogold labelling and SEM
SEM was used to visualize the nanoarchitecture of PA-hydrogel-stabilized cardiac tissue after decellularization (DECIPHER samples) and native cardiac tissue cryosections cut at 10 µm (Leica CM1950) from snap-frozen fresh young hearts embedded in OCT (Tissue-Tek, Sakura Finetek). For immunogold labelling, the samples were first fixed with 4% formaldehyde and rinsed with 1× PBS before blocking with 3% goat serum and 1% BSA for 30 min. Endogenous mouse IgG was blocked followed by incubation with a site-specific primary antibody. Samples were then rinsed with PBS + 0.1% Tween 20 and incubated with secondary antibody conjugated with 10-nm gold (Invitrogen) according to the supplier’s instructions. Immunogold-labelled samples were then rinsed and enhanced using Nanoprobes GoldEnhance EM Plus kit to improve the SEM visualization. Before SEM, all the samples were fixed with glutaraldehyde (Sigma-Aldrich) followed by dehydration using a series of ethanol exchanges of increasing concentration and dried using a Critical Point Dryer (Tousimis Autosamdri-815). The samples were coated with 8-nm platinum (for native tissue samples and unstained DECIPHER samples) or carbon (for immunogold samples) before being imaged using a Hitachi Regulus 8230 FE-SEM at 1–3-kV acceleration voltage and 8-mm working distance.
Nanoindentation
To obtain the Young modulus of PA hydrogels and PA-hydrogel-stabilized cardiac tissue slices pre- and post-DECIPHER, the samples were attached to glass-bottom Petri dishes with superglue. For native tissues, vibratome slices were prepared at 500 μm and attached to dishes with GLUture (World Precision Instruments, 503763). All the samples were then indented with the Optics11 Life Chiaro Nanoindenter using a spherical probe (k = 0.5 N m–1; tip radius, 50 μm). To minimize sample–probe adhesion, 1% Pluronic (Sigma-Aldrich) solution was used to coat the probe before the experiments. Calibration was carried out against a glass dish. Quasi-static matrix scans were performed with a 2-μm indentation depth and step sizes of 500 μm (native tissue, PA-stabilized tissue and DECIPHER samples) or 800 μm (plain PA hydrogel samples). The Young modulus of each data point was obtained from the Hertzian contact model with Poisson’s ratio ν = 0.5 in the Optics11 DataViewer software (v.2.7.0).
Dynamic mechanical analysis was used to measure the storage and loss moduli with a 2-μm indentation depth and 300-nm oscillation amplitude. The maximum indentation depth in the dynamic mechanical analysis was reached at 0.1 s, followed by 20-s relaxation, and then an oscillatory frequency sweep at 1, 2, 4 and 10 Hz was performed. Five periods of oscillation were carried out for all frequencies, except in the 10-Hz experiment, where ten periods were used to ensure a minimum oscillation time of 1 s. The storage modulus (E′) and loss modulus (E″) were calculated using the Optics11 DataViewer software.
Correlative confocal–AFM
AFM calibrations and measurements were performed after confocal imaging23. Briefly, a JPK NanoWizard 4 XP Bioscience AFM system with a BioMAT Workstation (Bruker) were used in conjunction with an upright Stellaris 8 laser scanning confocal microscope (Leica) equipped with a customized BioMAT shuttle stage, which enabled the precise positioning and alignment of the measured areas for the sequential confocal–AFM scans. A triangular cantilever (Nanoworld, Pyrex-Nitride Probe) with a nominal spring constant of 0.32 N m–1 with a pyramidal tip of 35° face half-angle were used. Calibration for deflection sensitivity and the spring constant was done under water with the contact-based calibration mode on a glass slide. The manufacturer’s calibration slide (Bruker) was used to optically align the confocal and AFM scan areas using the DirectOverlay function of the JPK NanoWizard data processing software (v.8.0.144), and minor alignment corrections were performed manually in post-processing.
The DECIPHER and native tissue samples were first stained with CNA35 without fixation (to provide live visualization of the collagen ECM) and imaged under the confocal microscope to obtain a three-dimensional stack and locate the regions of interest of the exposed ECM (collagen) fibres and the surrounding hydrogel (for DECIPHER). The samples were then transferred to the AFM system on the shuttle stage to perform surface topographic scans using the cells in the liquid quantitative imaging mode (force setpoint, 15–25 µN; indentation depth, 0.5–2 µm; Z speed, 25–40 µm; Z length, 4–5 µm; scan rate, 25–40 kHz; pixel time, 20 ms; x–y resolution: –312.50 nm per pixel). The scan sizes ranged in 25 × 25 µm2 to 50 × 50 µm2 with a resolution from 32 × 32 pixels to 80 × 80 pixels, respectively. After scanning, all the quantitative imaging images and force–distance curves were analysed in the JPK NanoWizard data processing software (v.8.0.144). The force–distance curves underwent baseline correction and smoothing before contact-point determination and fitting for Young’s moduli. The apparent Young’s modulus for each pixel in the quantitative imaging image was obtained by fitting a Sneddon contact model for the whole approach curve with ν = 0.5 to obtain the modulus maps. For quantification, three to five regions of interest with sizes of 4–6.25 µm2 were selected on the fibre and on the surrounding gel (for DECIPHER) to derive the regional average moduli.
Primary CF isolation and in vitro culture
The use of animals was approved by the Institutional Animal Care and Use Committee (protocol no. R22-0579), National University of Singapore. Mice are housed in individually ventilated cages with sex-matched littermates, 12-h light–dark cycles, ambient temperature (20–24 °C) and humidity (30%–70%), and ad libitum food and water supply. Primary young and aged CFs were extracted66 from female young (1–2 months) and aged (24 months) C57BL/6J murine hearts, respectively. Briefly, mouse hearts were perfused with cardiac digestion buffers by intraventricular injection. Three rounds of gravity settling were applied to separate cardiomyocyte and non-cardiomyocyte cells. Non-cardiomyocyte cells were seeded onto untreated tissue culture plastic and washed after 1 h to enrich for CFs. CFs were expanded in CF growth medium (Dulbecco’s modified Eagle’s medium/high glucose (Cytiva) supplemented with 15% fetal bovine serum (Gibco)), penicillin (100 U ml–1), streptomycin (100 µg ml–1, Nacalai Tesque), FGF2 (10 ng ml–1, Stemcell Technologies) and SB-431542 (5 µM, Targetmol).
All cells used in the in vitro studies were from passages 2 or 3. Before cell seeding, all the DECIPHER samples were immersed in sterile 1× PBS overnight, UV sterilized in a biosafety cabinet for 60 min and equilibrated in full maintenance media for 30 min. The isolated cells were seeded at 10,000 cells per well of a 12-well plate in 1 ml of culture media (Dulbecco’s modified Eagle’s medium + 1% fetal bovine serum + 1% pen/strep) onto DECIPHER samples and kept in a 37 °C, 5% CO2, humidified incubator for a culture period of 2 days. Cell culture supernatant was regularly tested negative for mycoplasma contamination using InvivoGen’s MycoStrip.
ICC
After culturing, cells were rinsed in sterile 1× PBS three times and then fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 15 min and blocked with 2% BSA for 30 min for ICC. For mouse-anti-mouse primary antibodies, 2% goat serum with 1% BSA was used instead as the blocking and staining buffer, with an extra step of blocking endogenous mouse IgG for 1 h at room temperature to reduce the background noise from the ECM. Samples were stained with primary antibodies (Supplementary Table 2) overnight at 4 °C in 2% BSA, rinsed three times in 1× PBS + 0.1% Tween 20, followed by secondary antibodies and CF Phalloidin for 1 h at room temperature diluted in 2% BSA. The samples were washed with PBS + 0.1% Tween 20 three times (5 min each) and stained with Hoechst 33342 in H2O for 10 min at room temperature before three rounds of water rinses and mounted with Fluoromount-G.
Low-input bulk RNA-seq
All the reagents are DNase/RNase/protease free and diluted with HyPure Molecular Biology Grade Water (Cytiva). Cells attached to the DECIPHER samples were lysed using TRIzol (Ambion) after scraping away the excess hydrogel region and kept under −80 °C pending RNA extraction. To further provide baseline (in vivo) control groups, freshly isolated young/aged primary CFs were directly lysed with TRIzol after extraction. Total RNA of the samples was extracted using a chloroform–isopropanol approach according to the supplier’s instructions. Briefly, chloroform was added to the lysed cell samples after thawing and vigorously mixed and centrifuged at 14,000g for 15 min before the supernatant was collected. RNA was then precipitated using cold isopropanol, collected by centrifugation at 14,000g for 15 min and washed twice with precooled 75% ethanol followed by centrifugation at 14,000g for 10 min. The RNA samples were air dried for 5–10 min before being dissolved in HyPure water and processed for quality control with TapeStation High Sensitivity RNA ScreenTape Analysis. Sample RNA with an RNA integrity number (RINe) larger than 8.5 (high quality) was applied to NEBNext Single Cell/Low Input RNA Library Prep Kit for Illumina for cDNA synthesis and sequence-ready library preparation according to the supplier’s instructions. Specifically, 20 ng of total RNA from each sample was used for cDNA synthesis and eight PCR cycles were used for cDNA amplification. The libraries were indexed with NEBNext Multiplex Oligos for Illumina (96 Unique Dual Index Primer Pairs Set 2) and submitted to NovogeneAIT Genomics (Singapore) for sequencing on the Illumina NovaSeq 6000 system.
Multiplex RT-qPCR
RNA extraction and quality control were performed as described in the RNA-seq section. Subsequently, a Reliance One-Step Multiplex RT-qPCR Supermix (Bio-Rad) was used to perform reverse transcription and qPCR by PrimePCR Probe Assays built in a Bio-Rad CFX96 Real-Time PCR Detection System. The thermal cycling protocol was set to the Bio-Rad product sheet default: 50 °C for 10 min (RT), 95 °C for 10 min (DNA polymerase activation and template denaturation) and 40 cycles of 95 °C for 10 s followed by 60 °C for 30 s (amplification). All the PrimePCR Probes used in this study were purchased from Bio-Rad (Supplementary Table 3), which had been optimally designed and validated for the multiplex qPCR supermix and experiments.
Imaging and analysis
The IHC and ICC samples were observed using a Nikon A1R scanning laser confocal microscope (×40 and ×60 water-immersion objectives, NIS Elements v. 5.30.05) or Oxford Instruments BC43 spinning-disc confocal microscope (×20 and ×40 air objectives, Fusion v. 2.3) and subsequently imported to Fiji. A Fiji macro, TWOMBLI, was used to quantify the ECM architectural changes post-decellularization and in ageing with the default parameters20. Confocal images from pre- and post-decellularized samples were analysed for lacunarity, endpoints, branch points, fractal dimension, alignment and hyphal growth unit, with a principal component analysis performed in GraphPad Prism v. 10. Confocal imaging was performed on Nile Blue-tagged samples from the sample surface and reconstructed using Imaris. LOX and NF-κB staining on DECIPHER and native tissue samples were quantified for their co-localization with collagen (CNA35) using Fiji JaCoP macro67. ICC samples stained with α-SMA, integrin α5, LOX, p53, TIMP3 and F-actin were analysed with CellProfiler for intensity and morphological metrics. Nucleus/cytosol localization ratio of YAP was quantified using CellProfiler (v.4.2.5) by identifying the fluorescence from a 30-pixel perinuclear ring as the cytosolic localization. Paxillin quantification was performed using a custom Fiji script in which focal adhesion sites were identified using auto local threshold (Bernsen) followed by particle analysis.
Bioinformatics analysis
For mass spectrometry data, the raw reads were obtained with FDR ≤ 1% and quantitative analysis was performed based on the peak intensity, peak area and liquid chromatography retention time of the samples, followed by a series of statistical analysis and quality control. The protein identification results were based on UniProt Protein Database (Mus musculus). Subsequent bioinformatics analysis and data visualization were processed using BGI’s Dr. Tom online analysis platform. Additional data visualization was performed using OriginPro 2024 and TBtools68.
RNA-seq reads were aligned to the mouse genome assembly mm39 (BioProject no. PRJNA20689) with STAR (v.2.7.8a). The aligned reads were quantified using HTSeq (0.11.0) with Ensembl transcripts release 105. The above computation was performed within the PartekFlow cloud environment with default parameters (v.10.0.23.0425). DEG analysis was done with the DESeq2 package69 (Bioconductor 3.17) and clusterProfiler package70,71 (Bioconductor 3.17) was used to perform the functional enrichment analysis. Cellinker31 was used to correlate the ligand–receptor interaction between ECM proteomics and CF RNA. Data visualization was performed using OriginPro 2024, GraphPad Prism 9, MATLAB R2023a and TBtools68.
Statistics and reproducibility
No statistical method was used to predetermine the sample size. Sample sizes were determined based on previous studies in similar fields. Unless otherwise noted, all the experiments were performed as triplicate independent experiments (N ≥ 3). Detailed sample sizes can be found in the respective figure captions. The number of cells analysed are reported in each figure with individual data points as well as in Supplementary Table 4. Data are expressed as the mean ± standard deviation (s.d.) or standard error of the mean (s.e.m.), with the sample size and applied statistical analysis specified in each figure caption. Statistical analyses were performed using GraphPad Prism v. 9 and v. 10. All the box graphs shown in the figures have the box range showing the interquartile data, centre of the box represents the median, ‘+’ mark represents the mean, and whiskers represent the minimum and maximum values. Representative IHC and ICC images shown in this study were successfully reproduced with similar results in three independent attempts. Other representative micrographs, such as Figs. 1c and 2a,b, were successfully reproduced with similar results for more than ten times throughout this study. No data were excluded from the analyses. In vitro experiments and samples used were randomly allocated. The investigators were blinded during data collection but not blinded during outcome assessment because the analyses were done objectively with automated scripts and codes.
Heat maps demonstrating the DEG analysis in the RNA-seq data were graphed by averaging the replicates, with the original (unaveraged) readings and dendrograms shown in Extended Data Fig. 4 (except in Fig. 3b where the original data are shown). The heat map in Fig. 3j is shown following the order of the other DEG heat maps, with the original clustering dendrogram shown in Extended Data Fig. 4a. The gene expression results for the DECIPHER samples are normalized to the young and aged groups of their respective in vivo mimic, that is, young CFs are normalized to the young ECM in a soft matrix (young CFs on SoftY), whereas aged CFs are normalized to the aged ECM in a stiff matrix (aged CFs on StiffA). The Ct (cycle threshold) values of RT-qPCR results were obtained via Bio-Rad CFX Maestro software (v.2.3) and analysed by the 2−ΔΔCt method72 against glyceraldehyde 3-phosphate dehydrogenase and the aforementioned in vivo mimic groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.